Chapter 2 Materials and Methods
2.1 Materials
2.1.1 Chemicals
| Chemical | Company | cat.-no. |
|---|---|---|
| Agarose | Roth | 6351.2 |
| Agar-Agar | Roth | 5210.3 |
| Ampicillin | Roth | K029.2 |
| ATP | Epicentre | E311K |
| Blocking Reagent | Roche | 11096176001 |
| BCIP | Fermentas | R0822 |
| CaCl | Roth | 886.1 |
| Calyculin | Sigma | 208851 |
| DAPI | Sigma | D9542 |
| DIG RNA Mix | Roche | 11277073910 |
| DMSO | Roth | 4720.2 |
| EtBr | Roth | 2218.3 |
| EtOH | Roth | 9065.3 |
| Formaldehyde | Roth | 7398.1 |
| Formamide | Roth | P040.1 |
| Glycerol | Roth | 3783.2 |
| IPTG | Thermo | R1171 |
| KCl | Roth | P017.1 |
| Low melting point Agarose | Roth | A9539 |
| Maleic Acid | Roth | 3810.3 |
| MgSO | Roth | T888.2 |
| MeOH | Roth | CP43.3 |
| MgCl | Roth | 2189.1 |
| NaCl | Roth | 9265.2 |
| NaHCO | Roth | 855.1 |
| NaOH | Roth | 6771.3 |
| NGS | Sigma | C6767 |
| p-Formaldehyd | Sigma | P6148 |
| Phenol Red | Sigma | P0290 |
| Propan-2-ol | VWR | 20842330 |
| Proteinase K | Roth | 7528.4 |
| PTU | Sigma | P7629 |
| Rockout | Sigma | 555553 |
| SSC | Roth | 1232.1 |
| SU5402 | CALBIOCHEM | 572630 |
| Torula RNA | Sigma | R6625 |
| Tricaine | Sigma | A5040 |
| Tris Base | Roth | 4855.2 |
| Triton-X100 | Roth | 3051.2 |
| Trizol | Ambion | 15596018 |
| Tween20 | Sigma | P1379 |
2.1.2 Solutions
| Solution | Company | cat.-no. |
|---|---|---|
| Cut Smart Buffer | NEB | B7204S |
| Generuler 100 bp | Thermo | SM0241 |
| Generuler 1kb | Thermo | SM0311 |
2.1.3 Antibodies
| Antibody | Company / Provider | concentration | cat.-no. |
|---|---|---|---|
| Anti-Digoxigenin | Roche | 1:200 | 11093274910 |
| Anti-GFP | Torrey Pines | 1:200 | - |
| Anti-TAZ (rabbit) | Cell Signaling Technology | 1:200 | D24E4 |
| Anti-ZO1 | Zymed | 1:200 | 33-9100 |
| Alexa Fluor488 | Invitrogen | 1:500 | 710369 |
| Alexa Fluor555 | Invitrogen | 1:500 | Z25005 |
2.1.4 Enzymes
| Enzyme | Company | cat.-no. |
|---|---|---|
| BtsCI | NEB | R0647 |
| DdeI | NEB | R0175 |
| HaeIII | NEB | R0108 |
| MnlII | NEB | R0163 |
| NlaIII | NEB | R0125 |
| NsiI-HF | NEB | R3127 |
| Phusion Polymerase | NEB | M0530L |
| Pronase | Sigma | P5147 |
| Ribolock | Thermo | EO0381 |
| RNase A | Quiagen | 1006657 |
| RNase H | NEB | M0297L |
| SP6 RNA Polymerase | Thermo | EP0131 |
| T4 Ligase | NEB | M0202T |
| T7 RNA Polymerase | Thermo | EP0111 |
| Taq DNA Polymerase | Invitrogen | 10342-020 |
| Taq DNA Polymerase | VWR | 733-1301 |
2.1.5 Molecular Biology Kits
| Kit | Company | cat.-no. |
|---|---|---|
| EdU Click-iT | Invitrogen | MP 10083 |
| mMessage mMachine Sp6 Polymerase | Invitrogen | AM1340 |
| PCR & Gel Clean-Up | Sigma | NA1020 |
| pGEM-T TA Cloning | Promega | A3600 |
| Superscript III cDNA Synthesis | Thermo | 18080051 |
| Wizard SV Gel and PCR Clean-Up | Promega | A9282 |
2.1.6 Buffers
| Buffer | |
|---|---|
| Blocking Reagent (BR) | 2% BR in maleic buffer + 5% serum |
| E3 | (52) |
| Hybridization buffer | 50% Formamide + 25 % 20x SSC + 50mg/mL Heparine + mQ |
| Maleic buffer | 250 mM maleic acid + 5M NaCl + 10% 0.1% Tween-20 + mQ |
| NTMT | 5 M NaCl + 1 M MgCl + 1 M Tris pH 9.5 + 10% Tween |
| PBS | 2.7 mM KCl + 12 mM HPO |
| PBST | PBS + 0.1% Tween20 |
| PBDT | PBS + 1% BSA + 1% DMSO + 0.3% Triton |
| PFA | 4% paraformaldehyde in PBS |
| P1 | 50 mM Tris-HCl pH 8.0 + 10 mM EDTA pH 8.0 + 100 µg/mL RNAse |
| P2 | 1M NaOH + 10 % (w/v) SDS |
| P3 | 3M KOAc pH 5.5 |
| TNT | 50 mM Tris-HCl pH 8.0 + 100 mM NaCl + 0.1% Tween-20 |
2.1.7 Zebrafish lines
| Allele | name | zfin |
|---|---|---|
| zf106Tg | cldnb:lyn-gfp | Tg(-8.0cldnb:LY-EGFP) |
| fu13Tg | cxcr4b(BAC):H2BRFP | TgBAC(cxcr4b:Hsa.HIST1H2BJ-RFP) |
| nns8Tg | atoh1a:Tom | Tg(atoh1a:dTomato)nns8 |
| fu50 | shroom3 | - |
| m1274Tg | hsp70:shr3v1FL-taqRFP | Tg1(hsp70l:shroom3-TagRFP) |
2.1.8 ISH probes
| Probe | Sequence |
|---|---|
| atoh1a | see (38) |
| deltaD | see (53) |
2.1.9 Morpholinos
| Probe | Sequence | Concentration |
|---|---|---|
| MoAtoh1a | see (38, 54) | 0.4 ng/mM |
| p53 | see (55) | 2 ng/mM |
2.1.10 Hardware
2.1.10.1 Mounting Stamp
An stl file for 3D printing can be found at github.com/KleinhansDa/3DModels
| Component | Company | cat.-no. |
|---|---|---|
| \(\mu\) dish | Ibidi | 81,218–200 |
| Stamp | - | - |
| Preparation needles | VWR | USBE5470 |
| Pasteur Pipettes | Roth | 4518 |
| Rubber / Silicone bulb | VWR | 612-2327 |
| Microtubes 2 mL | Sarstedt | 2691 |
| Heating block | PeqLab | HX2 |
| Microwave oven | Severin | MW7849 |
| Stereo microscope | Leica | M165FC |
| Transmitted Light Base | Leica | MDG36 |
| Countersunk screw DIN7991, 8 × 20 mm | Dresselhaus (Hornbach) | 7662389 |
| Superglue | UHU | 509141 |
2.1.10.2 Spinning Disc Microscopy
| Component | Company | Product | Specs |
|---|---|---|---|
| Microscope | Nikon | Eclipse Ti-E | fully motorized |
| PFS | Nikon | Perfect focus system | Z repositioning |
| XY-table | Merzhaeuser | XY motorized table | 1 \(\mu\)m accuracy |
| Piezo | Piezo Z-table | 300 \(\mu\)m scan range | |
| SD system | Yokogawa | CSU-W1 | 50 \(\mu\)m pattern |
| Laser | Laser Combiner | see table 2.12 | |
| FRAPPA | Revolution | FRAPPA | - |
| Borealis | Borealis | Borealis | flat field correction |
| sCMOS | Andor | ZYLA PLUS | 4.2Mpix; 82%QE |
| Immersion | Merzhaeuser | Liquid Dispenser | - |
| Lasers | Type | Power |
|---|---|---|
| 405 nm | diode | 100 mW |
| 445 nm | diode | 80 mW |
| 488 nm | DPSS | 100 mW |
| 561 nm | DPSS | 100 mW |
| 640 nm | diode | 100 mW |
| Objective | Company | Type | Immersion | N.A. | working distance |
|---|---|---|---|---|---|
| 10x | Nikon | CFI APO | air | 0.45 | 4.00 mm |
| 20X | Nikon | CFI APO | water | 0.95 | 0.95 mm |
| 40X | Nikon | CFI APO | water | 1.15 | 0.60 mm |
| 60X | Nikon | CFI APO | water | 1.20 | 0.30 mm |
2.1.10.3 Workstation
Statistical computation and image analysis were done on a Fujitsu Siemens (FS) Workstation CELSIUSM740 with the following hardware components…
| Component | Company | Product | Specs |
|---|---|---|---|
| CPU | Intel | Xeon E5-1660v4 | 3.2 GHz, 20MB, 8cores |
| RAM | Fujitsu | - | 4x16GB DDR4-2400 |
| Graphics | NVIDIA | Quadro M4000 | 8GB RAM |
2.1.11 Software
| Software | Version | web |
|---|---|---|
| Imagej FIJI | 1.48 | https://fiji.sc/ |
| R | 3.6.1 | https://cran.r-project.org/ |
| RStudio | 1.0.153 | https://www.rstudio.com/ |
| Ubuntu | 17.1 | https://www.ubuntu.com/ |
| Windows 10 Pro | 10.0.16299 | |
| Total Commander | 9.0 | http://ghisler.com/ |
2.2 Methods
2.2.1 Data Strategy and Analysis
Due to the history of Developmental Biology and the complexity of biological processes per se, the field heavily relies on image data. Since the advent of electronic imaging techniques11 scientific image data can be processed and analyzed in silico. To take advantage of
- live imaging, which (as compared to fixation techniques) conserves the cellular integrity and morphology while also offers the possibility of recording time-lapses
- the optically clear specimen and
- high throughput image analysis and state-of-the-art data science using algorithmic implementations,
the three following points were paid special attention to.
2.2.1.1 Sample Preparation
For fluorescence microscopy zebrafish embryos are usually immersed in a 1\(\%\) solution of low melting-point agarose (LMPA) and then oriented on an optical cover slip manually until the LMPA has solidified. This process allows to mount12 eight to ten embryos per dish. To make use of the high number of offspring, a single zebrafish female may lay, which rapidly leads to a sample number of more than 300, a new sample preparation technique was designed that allows for (1) a four to five - time increase in samples per dish (2) a facilitated navigation via a grid-like orientation through the samples and (3) an improved spatial orientation where the embryos body axes are aligned parallel to the optical Z-sections of the confocal microscope. For details, see Materials and Methods section 2.2.3.4 and Kleinhans et al., 2019 (56).
2.2.1.2 Imaging
Technically, speed and sensitivity are most important for live imaging. Considering these two parameters a light-sheet (57) fluorescence microscope (LSFM) would be the best fit. However, LSFMs also have several limitations. First, due to the sample preparation methods available, the number of samples that can be imaged at a time is highly restricted. Second, for subcellular resolution a high magnification is required, which is limited by working distances and third - for optimal image analysis a high signal-to-noise ratio (SNR) and numerical aperture (N.A.) is preferable. Therefore a spinning disc (58) system was chosen for most of the imaging. The system makes use of (1) an extra-large field of view (FOV) ideal for large specimen, (2) the possibility of a high degree of automation with state-of-the-art software and (3) a water dispensing system for long-term water immersion imaging. For details about the system see Materials section 2.1.10.2.
2.2.1.3 Data handling
After data acquisition and pre-processing, the image data was transferred from the microscope system to the labs main workstation. To uniquely identify each file and have them appear in a structured manner, a file-naming system was established after the following structure
Where stage would e.g. be 32hpf, group would be a genotype or drug treatment, id would be a positional identifier on an imaging dish like B1P0113 and date would be a date in the form of YYMMDD.
2.2.1.4 Image and Data Analysis
In order to be as objective and as high throughput as possible, almost all of the analyses performed for this study was solved either algorithmically or using convolutional neural networks (CNNs). Furthermore, to meet the terms and conditions of open science14 standards, all pipelines were implemented in open source software frameworks such as Fiji is just image J (FIJI) and R. For further information about training datasets, algorithms and versions used see Materials section 2.1.11.
2.2.2 Zebrafish
2.2.2.1 Husbandry
Zebrafish husbandry was maintained at the University of Frankfurt am Main. All legal procedures were followed while handling and maintaining zebrafish husbandry. All zebrafish lines used in and generated for this study are listed in Materials section 2.1.7.
2.2.2.2 Handling and rearing
In the afternoon preceding the embryo collection, 1 male and female were set up in crossing cages, physically separated by a transparent separator. Next day, before noon, separators were removed allowing fertilization. Fertilized eggs were then collected, sorted and reared in the well-defined culture medium E3 (Kimmel et.al. 1995, section 2.1.6) at 25\(^\circ\), 28.5\(^\circ\), or 30\(^\circ\)C depending on the experimental condition required.
To grow larvae to the adulthood, they were transferred to the system on day 5. Till day 12, larvae were fed Vinegar Eels, Paramecia, and caviar powder. After the 12 th day, water supply was started and fish were fed Brine Shrimp, Artemia, Paramecia and Vinegar Eels. Adult fish (>1 Month) were fed Artemia and the dry flakes.
2.2.2.3 Zebrafish fin clips
Adult fish were anesthetized with buffered Tricaine (1X, see section 2.1.6) until loss of motion. About 1/3 of the caudal fin was cut with a sterile scalpel in a sterile Petri Dish. Immediately the dissected fin was transferred to 100 \(\mu\)L of 50 mM NaOH. Fish were returned to system water and kept in 1L system water in single tanks with 200 \(\mu\)L of 0.01\(\%\) Methylene Blue.
2.2.2.4 Adult Genotyping
The clipped fins were digested for 1 h at 95\(^\circ\) C and neutralized subsequently with 10 \(\mu\)L of 1M Tris-HCl of (pH 9).
2.2.2.5 Embryo Genotyping
Single fixed/live embryos were denatured at 95\(^\circ\) C in 20 \(\mu\)L of 50 mM NaOH for 1 hour and neutralized by adding 2.5 \(\mu\)L of 1 M Tris-HCl (pH 9).
2.2.2.6 Zebrafish Euthanasia
Adult zebrafish were euthanised by an overdose of Tricaine in ice cold water so as to sacrifice them by hypothermia.
2.2.2.7 Fixation
Embryos and dechorionated larvae were fixed in 2 mL of 4\(\%\) PFA in 1X PBS overnight at 4\(^\circ\) C.
2.2.3 Wet lab
2.2.3.1 Sample preparation
For samples older than 24 hpf, embryos were grown in 1X PTU till desired stage and treated with 150 \(\mu\)L per 10 mL of 0.1 mg/mL Pronase for ~30 min.. Choria were removed by gentle pipetting with a 2 mL plastic pasteur pipette. To replace the Pronase solution with fresh E3, embryos were immobilized by anesthesia and collected in the center of the dish by gentle rotational movement. Then the medium was decanted by collecting the embryos at the corner bottom of the dish while taking care not to loose any. After, the dish was filled with fresh E3. This process was repeated three times.
For samples younger than 18s stage, embryos were treated with 150 \(\mu\)L per 10 mL of 0.1 mg/mL Pronase directly. Choria were removed the same way as for > 24 hpf embryos but when pouring away the Pronase solution, the dish was simultaneously and very carefully filled with fresh E3 again. Since younger embryos are more fragile and to avoid damage, they must be kept in solution constantly.
Fixation started at the desired stage in 4\(\%\) PFA in 0.1\(\%\) PBST in 2 mL tubes at 4\(^\circ\)C o.n.. The next day, samples were rinsed 3 times for ~5 min. in PBST and passed through a MeOH series of 25\(\%\) \(\rightarrow\) 50\(\%\) \(\rightarrow\) 75\(\%\) \(\rightarrow\) 100\(\%\) MeOH/PBST (V/V)). For permanent storage, samples were stored in 100\(\%\) MeOH at -20\(^\circ\)C.
2.2.3.2 In Situ Hybridization
Samples were prepared after the method described in section 2.2.3.1.
2.2.3.2.1 1. Permeabilisation & Probe Hybridization
Permeabilisation \(\rightarrow\) without shaking
Samples were rehydrated in an inverse MeOH series of 75\(\%\) \(\rightarrow\) 50\(\%\) \(\rightarrow\) 25\(\%\) \(\rightarrow\) 0\(\%\) PBST and washed again for fice min. two times in pure PBST. Finally, samples were digested in 10 \(\mu\)g/mL Proteinase K according to table 2.16.
| Stage | 0.6 s | 7 s | 18 s | 24 hpf | 32 hpf | 36 hpf | 42 hpf | 48 hpf | 72 hpf |
|---|---|---|---|---|---|---|---|---|---|
| min. | 0 | 4 | 6 | 15 | 30 | 40 | 50 | 60 | 60 |
Samples were rinsed again two times in PBST and post-fixated in 4\(\%\) PFA at 4\(^\circ\)C for > 30 min. Samples were washed again for 5 min. three times in PBST.
Probe Hybridisation \(\rightarrow\) all steps at 60\(^\circ\)C, except stated differently
Samples were pre-hybridized in 350 \(\mu\)L of hybridization buffer (section 2.1.6) for 1 - 8 h. Just before detection probe treatment, the probe was denatured at 80\(^\circ\)C in 1:200 of hybridization buffer. Subsequently, hybridization buffer was taken off the samples and replaced with the heated probe. Finally, samples were incubated o.n. at a desired temperature (around 65\(^\circ\)C).
2.2.3.2.2 2. Probe removal & Antibody incubation
The next day the probe got collected and stored at -20\(^\circ\)C for re-use. Washing took place at the same temperature as hybridization (from step 1) To keep solutions at temperature a Thermo-Block was used. For the washing series the samples were first washed one time for 20 min. in hybridization buffer, then two times for 30 min. in 50\(\%\) Formamide and one time for 20 min. in 25\(\%\) Formamide. Then two times for 15 min. in 2X SSCT and two times for 30 min. in 0.2X SSCT. Finally, one time for 5 min. in TNT.
To reduce noise and increase specific signal strength, the samples were treated with blocking solution (section 2.1.6) for 1 - 8 h in 350 \(\mu\)L of 2% BR/TNT at room temperature (RT). Afterwards the samples were incubated in 100 \(\mu\)L Anti-Digoxigenin diluted in NTMT buffer (1/4000 (V/V)) in 2\(\%\) BR/TNT for 2 h at RT or o.n. at 4\(^\circ\)C.
2.2.3.2.3 3. Probe detection
First, the samples were washed six times for ~20 min. (or one wash o.n.) in TNT and two times for ~ 5min. in NTMT. After washing, color staining was performed with 4.5 NBT \(\mu\)L + 3.5 \(\mu\)L BCIP per mL NTMT in the dark and at RT without shaking (in a drawer) for 2 - 8 h, regularly checking the progression of the reaction. As soon as an appropriate degree of color intensity on the target site was achieved (up to two days), the samples were again washed three times in PBST.
Afterwards the samples were either prepared for immunostaining or imaging. For permanent storage samples were kept in 50\(\%\) Glycerol at 4\(^\circ\)C.
2.2.3.3 Immuno staining
Samples were prepared after the method described in section 2.2.3.1.
First, samples were blocked in 2\(\%\) Goat Serum / PBDT (V/V) for 30 min.. For protein target site detection, a primary antibody (150 \(\mu\)L of 2% NGS / PBDT (V/V)) was incubated for ~2 h at RT or o.n. at 4\(^\circ\)C. Samples were washed for 2 h in PBDT while changing the solution 5 - 6 times. To stain the now bound primary antibody, a secondary antibody (150 \(\mu\)L of 2% NGS / PBDT (V/V)) was incubated for 2 h at RT or o.n. at 4\(^\circ\)C. Samples were washed for 2 h in PBDT while changing the solution 5 - 6 times.
2.2.3.4 Mounting
For live microscopy zebrafish embryos are usually immersed in a 1\(\%\) solution of low melting-point Agarose (LMPA) solution and then oriented on an optical cover slip manually until the LMPA has solidified. This process allows to mount eight to ten embryos per dish.
To take advantage of the high number of offspring a single zebrafish female may lay, a new sample preparation technique was designed that allows for
- a four to five times increase in samples per dish
- a facilitated navigation via a grid-like orientation through the samples and
- an improved spatial orientation where the embryos body axes are aligned parallel to the optical Z-sections of the confocal microscope.
A detailed protocol can be found under section 3.1.1
2.2.4 Dry lab
2.2.4.1 Image J macros
Three IJ macros have been developed to facilitate image analysis and make results more reproducible. Each of them is specifically designed for input of LL and pLLP images of the cldnb:lyn-gfp transgenic line.
Hence, they are called anaLLzr …
- 2D - analysis of Z-projected images of the LL at end of migration
- 2DT - analysis of Z-projected images of the LL during migration
- 3D - analysis of 3D image stacks of the pLLP at a given timepoint
Since their development was an integral part of my PhD work, the description of the macros can be found in the results part in section section 3.1.
2.2.4.2 Proliferation Analysis
The basic principle is based on work done by Laguerre et al., 2009 (34). For registration of mitotic events an IJ manual tracking tool was used that allows to track an image feature through a stack of images creating tracks as it progresses through volume / time (‘MTrackJ’(59)).
For mounting the embryo, the procedure described in section 2.2.3.4 was used. Nuclei were visualized in a TgBAC(cxcr4b:H2B-RFP) transgenic line. After Z-projection of volumetric timelapses, mitotic events were tracked in each CC and the pLLP. Afterwards the data was exported as one table per embryo and processed by counting mitoses per pLLP / CC / total CC mitoses. Figure 2.1 shows an exemplary track for the data analyzed.

Figure 2.1: Tracking of mitotic events. T1-T3 show consequetive timepoints of a single nucleus.
2.2.4.3 Apical Index
2.2.4.3.1 Rationale
The earliest attempt found for indexing AC can be found in a study published by Lee et al(60) where they were interested in the ‘apical index’ (A.I.) of bottle cells during X.laevis gastrulation (figure 2.2 Lee). Another example for measuring AC is the apical constriction index (A.C.I., figure 2.2 Harding) for the cells of the D.rerio lateral line primordium (pLLP), which can be found in a study from 2012 where it was shown that FGFr-Ras-MAPK signaling is required for Rock2a localization and AC (61, 62).
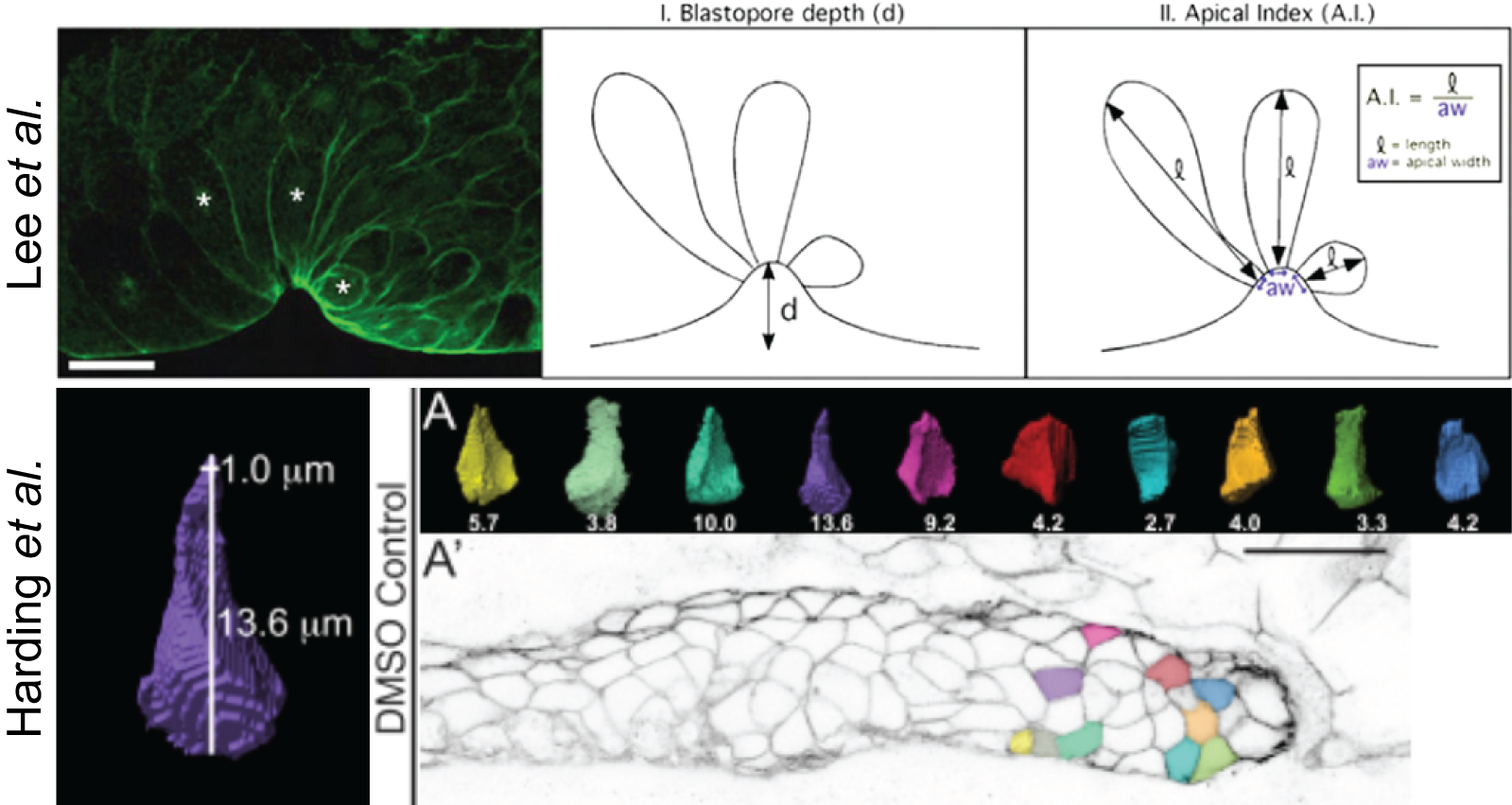
Figure 2.2: A.I. indeces in the literature. Lee A.I. of X.leavis bottle cells measured in 2D. Harding A.I. of D. rerio pLLP cells measured in 3D.
In these two publications, the way they measure A.I. (60) and ACI (62) respectively, does not differ and is the ratio of lateral height over apical width.
\[\mathbf{ACI} = \frac{lateral\;height\;[\mu m]}{apical\;width\;[\mu m]}\]
We found two principal weaknesses of applying this ratio to the cells of the pLLP. First, it does not respect the independence of lateral height to AC. Second, it does not differentiate between constriction along the anterio-posterior (AP) or the dorso-ventral (DV) axis. Third, it actually represents the A.I. rather than the apical constriction index.
2.2.4.3.1.1 Parameter definition
To obtain a precise and biologically meaningful way to quantify AC, first a couple of definitions had to be made.
2.2.4.3.1.2 Adaption for variation in lateral height
To test different A.I. conditions, an apically constricted cell can be approximated by modeling a tetrahedron. For example, shrinking or enlarging a cell symmetrically should not affect the A.I.. As described by Harding(2014)(62), the apical width of a cell is measured first by manual 3-D reconstruction, second manual re-orientation, and third by going 1 \(\mu\)m from the apical tip into the cell (from now on referred to as \(\Delta\)ap, 2.3B). Finally, apical width is the total width of the 2D object in the respective volume.
If \(\Delta\)ap is a constant, the A.I. in a symmetrically enlarged cell increases from e.g. ~15 to ~23, since apical width stays the same but lateral height increases (compare figure 2.3A to A’). On the contrary, if \(\Delta\)ap is adjusted relative to a cells lateral height, e.g. by percentage, the A.I. in a symmetrically enlarged cell stays the same (compare figure 2.3A to A’’).
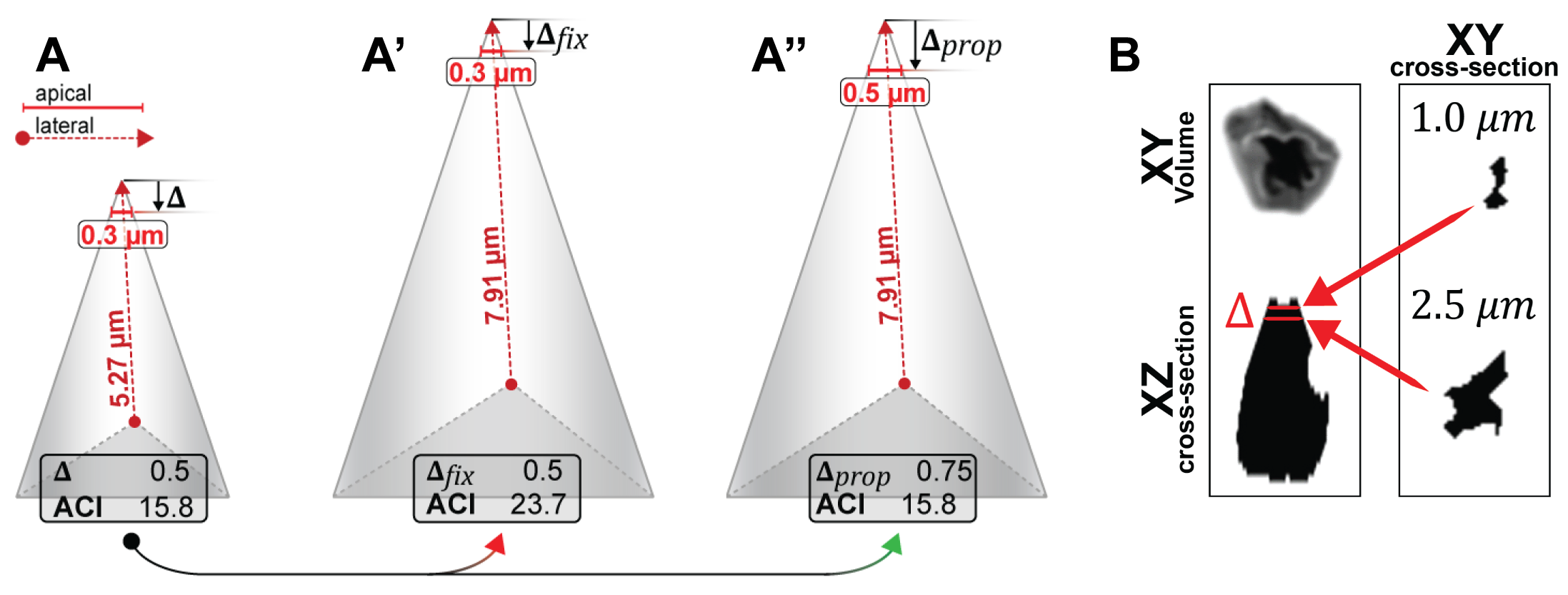
Figure 2.3: Different ways to quantify the apical index. A-A’’ A.I. Cell Models. A’ and A’’ show cells that are symmetrically increased versions of A. While in A’, constant delta was used, in A’’ delta is proportional to the lateral height. B Illustrating delta ap. (left) apically constricted cells volume rendered in XY (top) and as a lateral cross-section in X-Z (bottom). (right) 2-D area as seen at \(\Delta\)ap of 1 or 2.5 \(\mu\)m.
Therefore the measurement for apical width has to be relative to lateral height.
\[\mathbf{ACI} = \frac{lateral\;height\;[\mu m]}{relative\;apical\;width\;[\mu m]}\]
2.2.4.3.1.3 Adaption for tissue polarization
Organs develop in a 3-D space and are polarized along each axis. AC usually describes a 2-D morphogenetic movement towards a center along the X-Y axes. However, the constriction movements along X and Y might be independent of one another. This could mean that they happen at different speeds, or that one is absent. As a result, the tissue would look less radially (figure 2.4B) constricted, but more constricted along one particular axis (anisotropic). In order to separate those two AC dimensions, the A.I. can be calculated for the anterio-posterior and for the dorso-ventral axis (figure 2.4, horizontal vs. vertical).
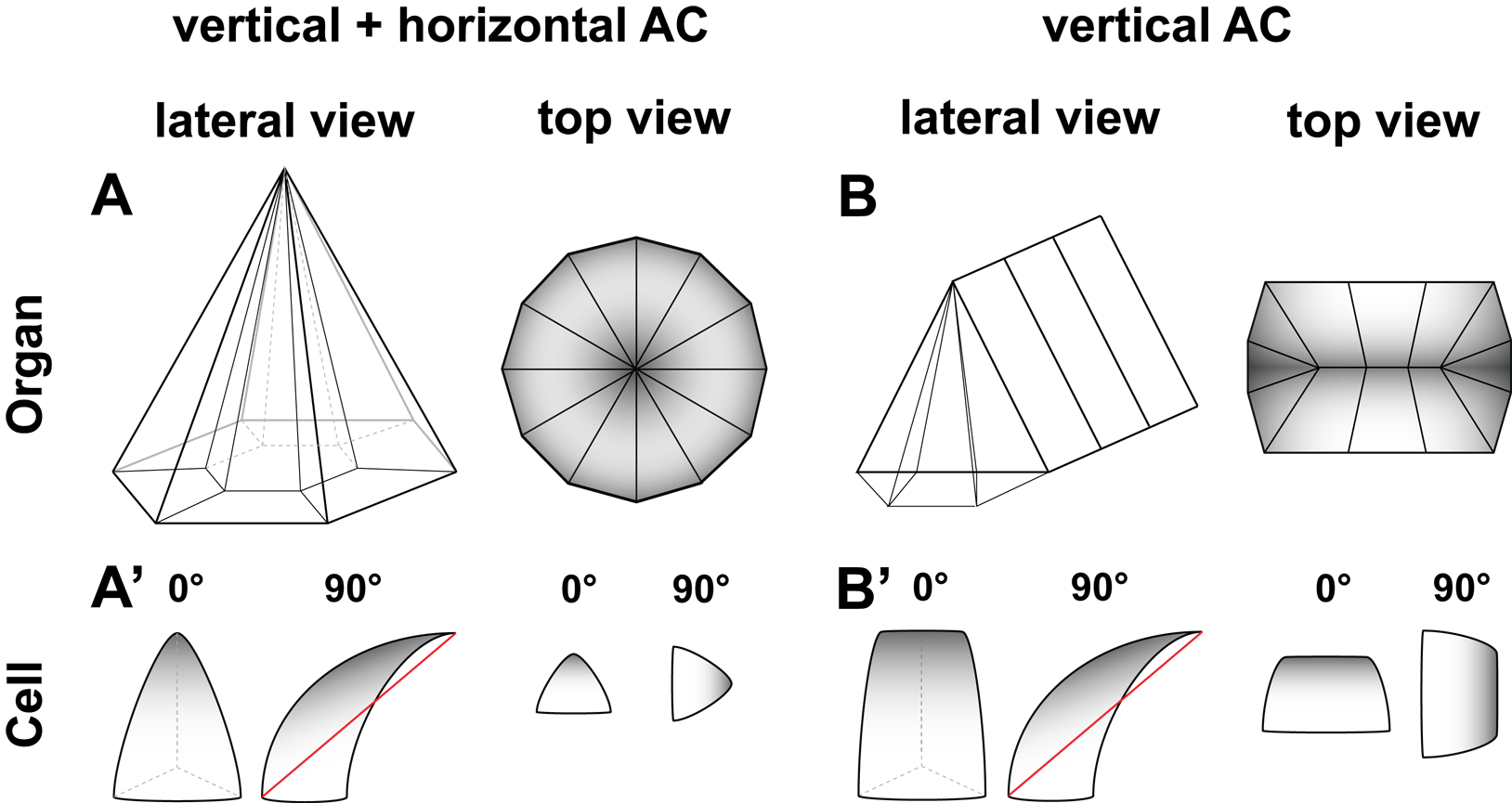
Figure 2.4: Schematic AC along the A-P and D-V axis. A shows a A-P and D-V constricted cluster of cells. B shows a D-V constricted cluster of cells.
By fitting an ellipsoid to the area taken at \(\mathrm{\Delta}\)ap, one will obtain the following parameters (figure 2.5).
- Length of Major axis
- indicates the level of constriction along the less constricted axis
- Length of Minor axis
- indicates the level of constriction along the most constricted axis
- Angle of Major from 0\(^\circ\)
- indicates the orientation of the long, less-constricted axis: If the angle is close to 0\(^\circ\), the long axis of the apical area is parallel to the AP axis (the cell is constricted along the DV axis). If the angle is close to 90\(^\circ\), the long axis of the apical area is parallel to the DV axis (the cell is constricted along the AP axis).
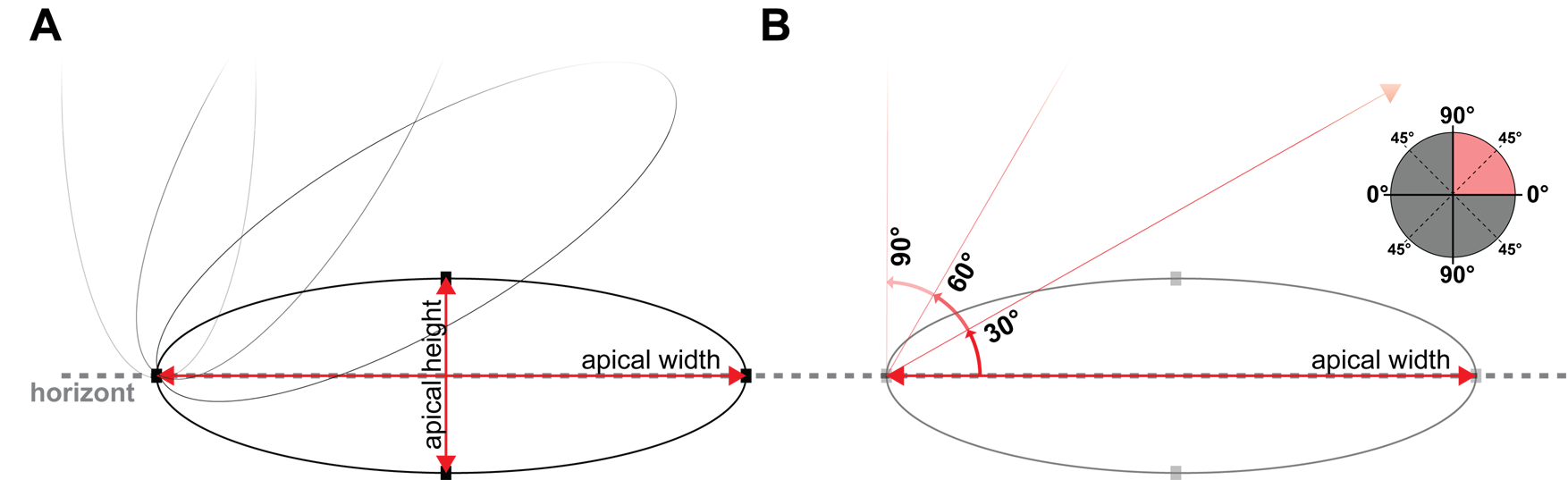
Figure 2.5: Scheme of ellipsoid measures. A shows the major axis as apical width and the minor axis as apical height. B shows the angular displacement from the horizon in steps of 30\(^\circ\).
2.2.4.3.1.4 Measurement definition
The two dimensions of A.I. indices can therefore be defined as the following ratios…
2.2.4.3.2 Measurements
As a proof of principle of the definitions stated in the previous section we compare our results to previously published results from Harding et al. (61) who, as a control, measured apical constriction in embryos treated with an FGF inhibitor (SU5402) and their DMSO controls.
2.2.4.3.2.1 Single cell measurements
Each geometric object has a centroid coordinate in X and Y (and Z) which is represented as the mean of all X or Y coordinates within the object. In figure 2.3, centroid coordinates in X and Y are used to plot the cells as points in the X-Y plane. Additionally, each point is colored for the A.I. value (high values are dark red - red, middle values are green, low values are cyan - blue).
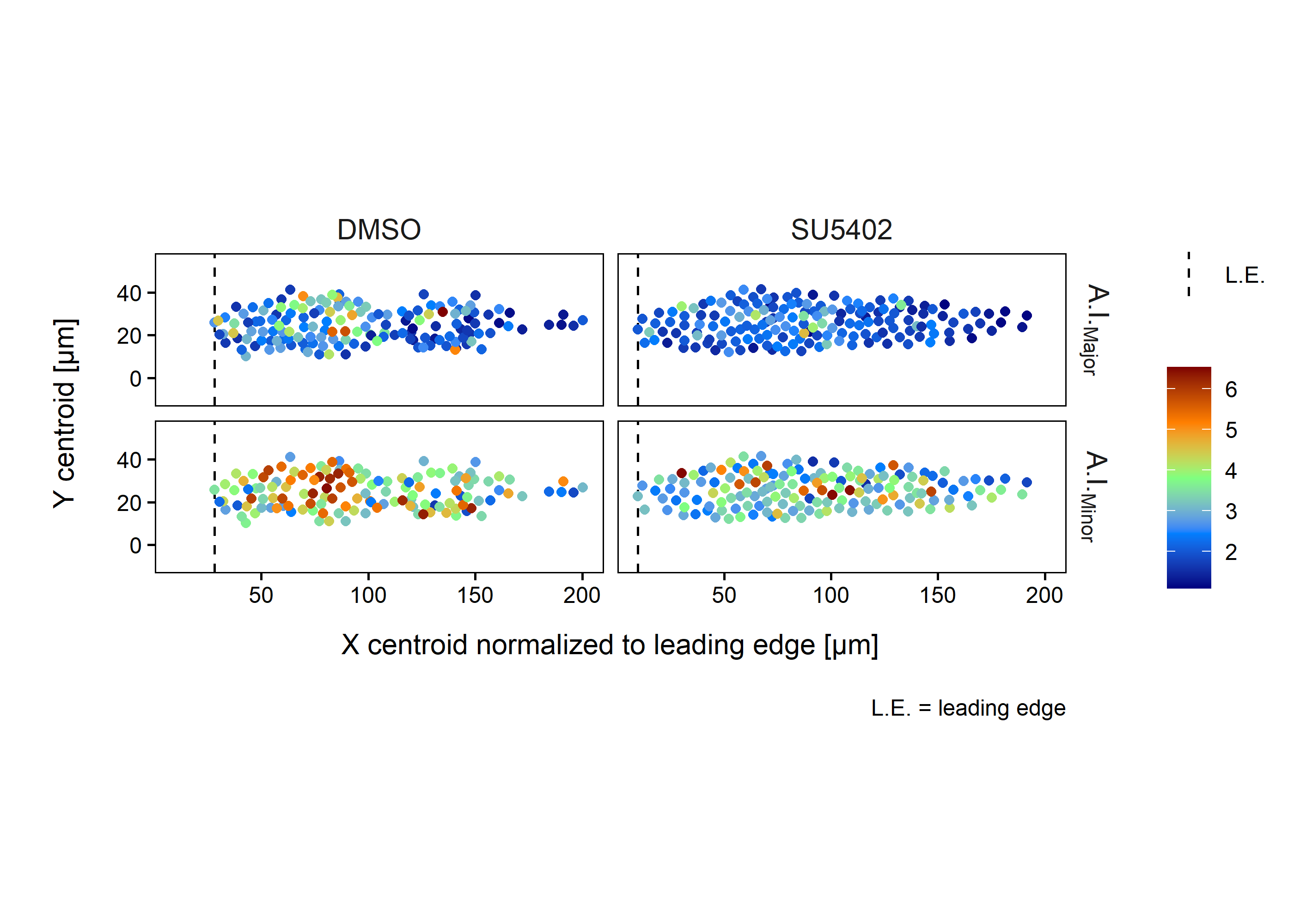
Figure 2.6: A.I. single cell measurements
Harding et al. (62) were using a constant \(\mathrm{\Delta}\)ap to measure the apical width, which we have shown to be incorrect in certain cases. In their study they found that certain mean A.C.I. values in the DMSO go as high as 15 (figure 2.7), which might be related to this (see figure 2.3). By measuring apical width at a relative \(\mathrm{\Delta}\)ap and taking into account all pLLP cells of the two exemplary pLLPs shown in figure 2.3, we measure a mean difference in the Major of 0.53 and 1.11 in the Minor.
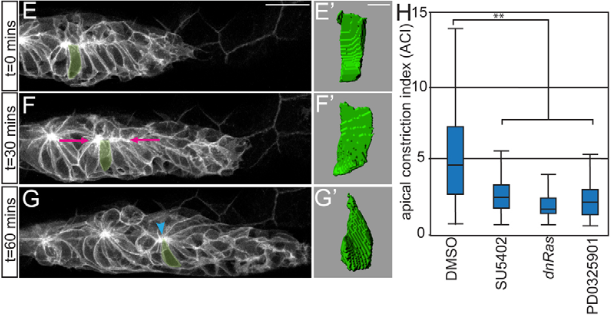
Figure 2.7: A.I. indices by Harding et al. E-G’ 3-D reconstructions of the highlighted cell. H A.C.I.s for embryos treated with DMSO, SU5402, PD0325901 or following induction of hsp70:dn-Ras. (n = 180 cells / N = 6 embryos).
2.2.4.3.2.2 Angle densities
To check whether there is a bias in orientation of the apical width, the angle measurements 2.5 can be shown as a function of density along X (figure 2.8A).
Interestingly the results indicate that there is less of a difference for the MajorAngle at angles bigger than 15-20\(^\circ\). This would mean that the apical surface of the cells in SU5402 treated embryos is more strongly oriented along the horizontal antero-posterior axis.
2.2.4.3.2.3 ACI magnitude at different angles
Now, to get an idea of the magnitude of constriction relative to the orientation of the cell (angle to the horizontal), the A.I.Major/Minor can be shown as a function of the MajorAngle (figure 2.8B-B’).
Since AC is a 3-D morphogenetic process and since cells in a wild type pLLP are mostly radially organized, it does make sense to look at AC from more than just one perspective. Here we propose to separate the A.I. into an antero-posterior and a dorso-ventral dimension.
While for the control (DMSO treated) embryo the distribution of the cells Major Angles seem to be mostly uniform, for the SU5402 treated embryo there is an accumulation of lower Major Angles. This means that cells in SU5402 treated embryos are more oriented along the horizontal (anterior - posterior) axis.
Interestingly there does not seem to be much of a difference in A.I.Major (figure 2.8B), which can also be shown by the mean values which are at 2.6 for the DMSO control and at 2.1 for the SU5402 treated condition.
For the A.I.Minor (figure 2.8B’) the means are 4.7 for the DMSO control and 3.6 for SU5402. The base constriction for both, DMSO and SU5402 is at around 3.6, however there is a peak at around 40 - 60\(^\circ\) in the DMSO control where cells are most constricted having a maximum A.I. at 15.8. This indicates that for the Minor, cells in that range of angles are more constricted than cells oriented in different directions.
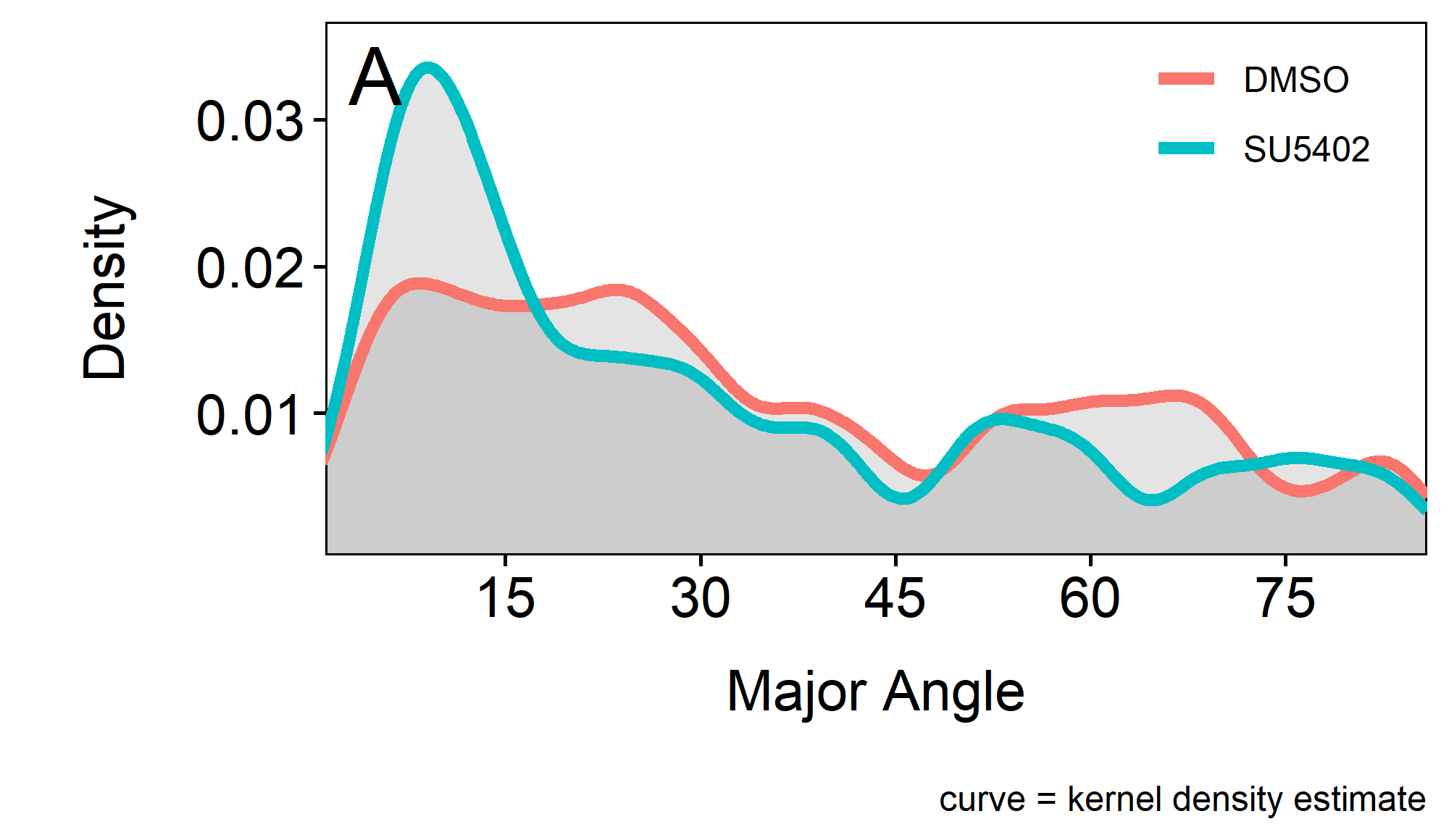
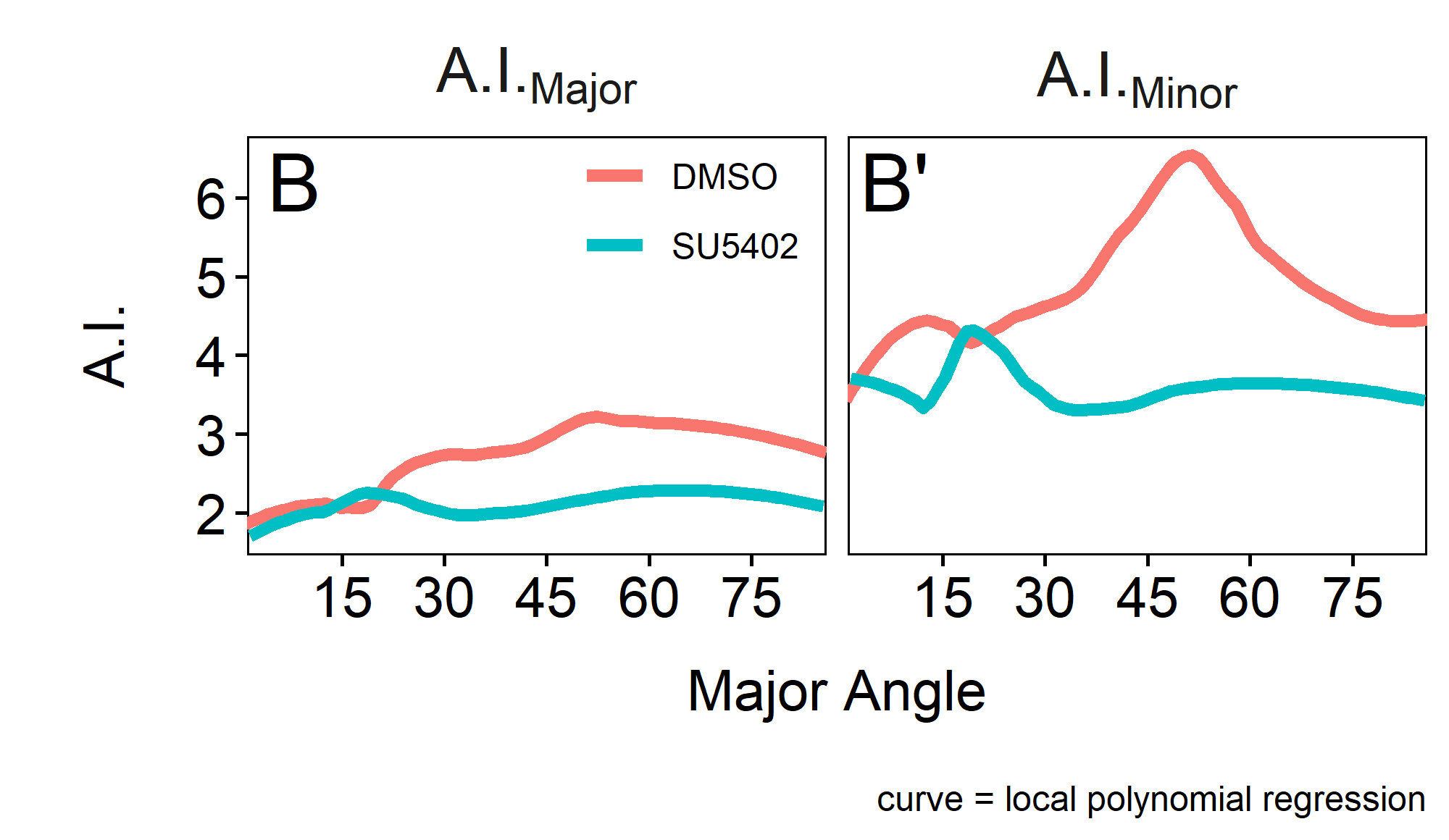
Figure 2.8: A.I.Major / A.I.Minor over MajorAngle
2.3 Ground Truth
Analyzing images and extracting quantitative measurements can be a tedious task, especially when the analysis becomes more complex. Fortunately, there are ways to automate image analysis by using either machine learning approaches or by tailoring hand-crafted algorithms in an image analysis software tool like e.g. ImageJ(63). The main advantages of doing so are to…
- be independent of confirmation bias
- make the analysis more robust against oversight
- increase the statistical power by increasing the number of data points(64)
- increase effect size by increasing the measurement accuracy(64)
However, to ensure the measurements taken by a tailored or trained algorithm are meaningful, they must be compared to a ground truth dataset which again describes a general measure of algorithmic quality performance(65).
2.3.1 Cluster Analysis
The anaLLzR2D algorithm was designed for semi-automatic cell cluster detection in the cldnb:lyn-gfp transgenic line and optional nuclei counting in a second DAPI-labeled channel within the Regions of Interest (ROIs) derived from the cell cluster detection.
2.3.1.1 Design
To assess the quality of the anaLLzR2D algorithm for nuclei detection the ground truth was designed as follows.
2.3.1.1.1 Model
- each Cell Cluster (CC) consists of a number of objects (cells)
- each object is part of the respective CC and defines one cell entity
- each object is determined via a fluorescent nucleus label
- embryos are mounted within a 3D mold (section 2.2.3.4) to reduce noise
| XY scale | 0.32 px/\(\mu\)m |
| Z-spacing | 4 \(\mu\)m |
| Camera | Rolera |
2.3.1.1.2 Training & test data
The training set consists of three randomly picked wildtype pLLs. For each the algorithm was run with standard parameters. Cell cluster ROIs and nuclei multi-point labels were edited manually. To test the algorithm, it is run at different nuclei detection thresholds on the same image data.
2.3.2 Morphometric analysis
The anallzr3D algorithm was designed for fully automated, single cell volume segmentation in the cldnb:lyn-gfp transgenic line. In addition to 3D cellular metrics, it offers Apical Constriction measurement of each cell.
2.3.2.1 Design
To assess the quality of the anaLLzr3D algorithm the ground truth was modeled as follows.
2.3.2.1.1 Model
- each pLLP consists of a number of objects (cells)
- each object is part of the pLLP and defines one cell entity
- cell boundaries are determined via the transgene cldnb:lyn-gfp +/+
- embryos are imaged live to conserve signal and membrane integrity
- embryos are mounted within a 3D mold for improved 3D alignment
| Exposure time | 100 ms |
| Laser intensity | 100\(\%\) / 9.3 mW |
| Objective | 40X; CFI APO LWD WI; N.A. = 1.15, W.D. = 0.60 mm |
| Tube lens | 1.0X |
| Z-spacing | 0.4 - 0.5 \(\mu\)m |
| Camera | sCMOS; 4.2 M.Pix; 82\(\%\) Q.E. |
| SD system | Yokogawa CSU - W1; 50 \(\mu\)m pattern |
| Piezo | Piezo Z-table; 300 \(\mu\)m scan range |
2.3.2.1.2 Training & test data
The training set consists of three randomly picked wildtype pLLPs. For each the algorithm is run with no filters (X, Y, Z border objects, size) and a minimum segmentation threshold. Afterwards the segmentation result is corrected manually for over- and under-segmentation and objects that are not part of the pLLP.
To test the algorithm it is run at different segmentation thresholds on the same image data.
2.4 Image Data Sets
Summaries of Image datasets. Pairs describe the number of parent pairs I harvested eggs from. Stage describes the time I waited for the parent pairs to mate and lay eggs. Since pair #1 might have laid their eggs earlier than pair #2, those batches would be some time apart in their staging. Stamp describes the version of the stamp I used, where the main difference between version 4 and 5 are more wells added and some minor upgrades in well-design.
| Crossings | Pairs | 4 |
| Transgenes | cldnb:lyn-gfp +/? | |
| Mutation | shroom3 | |
| Staging | 60 min. | |
| Mounting | Fixation | 4\(\%\) PFA o.N. |
| Agarose | 1\(\%\) LMPA | |
| Imaging | Magnification | 25X WI + 1.0x zoom |
| Channels | 488 nm | |
| Z-Stack | 2.5 \(\mu\)m Z-spacing; 110 \(\mu\)m stack size; 12*X large image |
| Crossings | Pairs | 4 |
| Transgenes | cldnb:lyn-gfp +/? | |
| Mutation | shroom3 | |
| Staging | 30 min. | |
| Mounting | Protocol | Kleinhans \(\textit{et al.}\), 2019 |
| Agarose | 0.5\(\%\) LMPA + 20\(\%\) Tricaine (V/V\(\%\)) | |
| stamp | version 4A | |
| Imaging | Magnification | 40X objective + 1.0x zoom |
| Camera | Binning 1x1; Gain 1; Exposure 100 ms | |
| Channels | 488 nm (100\(\%\)) | |
| Z-Stack | 0.4 \(\mu\)m Z-spacing |
| Crossings | Pairs | 6 |
| Transgenes | cldnb:lyn-gfp +/?; cxcr4b(BAC):H2BRFP +/0 | |
| Mutation | shroom3 | |
| Staging | 30 min. | |
| Mounting | Protocol | Kleinhans \(\textit{et al.}\), 2019 |
| Agarose | 0.3\(\%\) LMPA + 20\(\%\) Tricaine (V/V\(\%\)) | |
| stamp | version 4A | |
| Imaging | Magnification | 20X + 1.5x zoom |
| Camera | Binning 2x2; Gain 4; Exposure 35 ms; full FOV*150 \(\mu\)m | |
| Channels | 651 nm (25\(\%\)) | |
| Z-Stack | 2.5 \(\mu\)m Z-spacing; 110 \(\mu\)m stack size; 2*X large image | |
| Time | 20 h / 7 min. interval / start ~ 2 p.m. |
| Crossings | Pairs | six |
| Transgenes | cldnb:lyn-gfp +/? | |
| Mutation | shroom3 | |
| Staging | 30 min. | |
| Mounting | Protocol | Kleinhans \(\textit{et al.}\), 2019 |
| Agarose | 0.3\(\%\) LMPA + 20\(\%\) Tricaine (V/V\(\%\)) | |
| stamp | version 5A | |
| Imaging | Magnification | 20X + 1.5x zoom |
| Camera | Binning 2x2; Gain 4; Exposure 20 ms; full FOV*150 \(\mu\)m | |
| Channels | 488 nm (25\(\%\)) | |
| Positions | 36 | |
| Z-Stack | 3 \(\mu\)m Z-spacing; 100 \(\mu\)m stack size; 3*X large image | |
| Time | 20 h / 10 min. interval / start ~ 2 p.m. |
| Crossings | Pairs | four |
| Transgenes | cldnb:lyn-gfp +/?; atoh1a:Tom +/0 | |
| Mutation | shroom3 | |
| Staging | 30 min. | |
| Mounting | Protocol | Kleinhans \(\textit{et al.}\), 2019 |
| Agarose | 0.3\(\%\) LMPA + 20\(\%\) Tricaine (V/V\(\%\)) | |
| stamp | version 5A | |
| Imaging | Magnification | 20X + 1.0x zoom |
| Camera | Binning 2x2; Gain 4; full FOV*150 \(\mu\)m | |
| Channels | 488 nm (Int: 20\(\%\); Exp: 25 ms) | |
| Channels | 561 nm (Int: 30\(\%\); Exp: 50 ms) | |
| Positions | 36 | |
| Z-Stack | 2.5 \(\mu\)m Z-spacing; 100 \(\mu\)m stack size; 2*X large image | |
| Time | 20 h / 20 min. interval / start ~ 2 p.m. |
e.g. photomultipliers or charge-coupled devices↩︎
the process of embedding the samples in agarose↩︎
Where B stands for a batch, that is if multiple dishes were imaged and P stands for the position within a single batch↩︎
“movement to make scientific research […] and its dissemination accessible to all levels of an inquiring society” – Wikipedia/en/Open_science↩︎